|
COVID-19 RSS Patient Statement
“RSS/SRS children should be at no additional risk of contracting COVID-19 compared to the general pediatric population. They may be at a higher risk for complications once they have contracted the virus if they have additional risk factors such as asthma, preemie lung disease, feeding failure or other medical conditions. It is extremely important to continue to follow the current SRS Consensus Statement recommendations for the treatment of illness in RSS/SRS children [or SGA children with large head-size-to-body]. Please refer to the SRS Consensus Statement and the "Emergency Treatment Guidelines" document provided herewith. In case of acute illness, treat quickly and do not be afraid to go to the pediatric emergency room even with this COVID-19 pandemic. Emergency care for these patients should not be delayed."
*Drs. Irène Netchine, I. Karen Temple, Madeleine D. Harbison, Gerhard Binder, Justin H. Davies, Deborah J.G. Mackay, Oluwakemi Lokulo-Sodipe, Emma L. Wakeling, Thomas Eggermann
|
In October 2016, the Russell-Silver Syndrome International Consensus Statement was published. This comprehensive document, written by 36 international specialists in Russell-Silver syndrome, includes all of the diagnosis and treatment care recommendations voted on and approved by these specialists. This is a "must have" document for every medical professional caring for an individual with RSS. This document may be difficult to read for many non-medical individuals, so we now have an abbreviated, condensed version rewritten in more layman language -- the perfect solution for families and individuals managing RSS. Choose the full-length version, the condensed version ("Highlights and Summary", perfect for families and individuals with RSS), or the Spanish, Greek, French, Portuguese or Dutch version by clicking either option below.
KEY QUESTIONS & ANSWERS
-
What is Russell-Silver syndrome?
-
How is Russell-Silver syndrome (RSS) diagnosed?
-
What is the free RSS screening that MAGIC offers?
-
What is the typical RSS phenotype (physical characteristics)?
-
Are the cognitive abilities of RSS children within the normal range?
-
What should we do first if we think our child has Russell-Silver syndrome?
-
Why are RSS/SGA children at high risk for fasting hypoglycemia and ketonuria? And what preventative steps can we take?
-
What are the different treatments needed for Russell-Silver syndrome?
-
Do we need to improve our RSS child’s weight? What is an appropriate BMI for our child?
-
Does an RSS child’s delayed bone age mean more growing time later?
-
Do RSS children have early puberty?
-
Can my RSS child’s height be improved?
-
What are the different benefits and risks of growth hormone therapy (GHT) other than improving height?
-
Is a GH stimulation test needed for insurance coverage of GHT for RSS/SGA children? Are most RSS/SGA children growth hormone deficient (GHD)?
-
What are some tips in getting growth hormone therapy covered by insurance?
-
What are the different factors that can affect or improve the response to growth hormone therapy?
-
Are there other health issues that an individual with RSS may face in adulthood?
WHAT IS RUSSELL-SILVER SYNDROME
Russell-Silver syndrome (or Silver-Russell syndrome) is a rare genetic disorder characterized by delayed growth in-utero (IUGR) that spares head growth (meaning the newborn has a head size that is large for his body) and ongoing postnatal growth failure. This disorder includes feeding difficulties and/or low BMI, dysmorphic features including a protruding forehead, and frequently body asymmetry (hemihypotrophy). The true incidence is unknown but is estimated at 1 per every 35,000 – 100,000 live births.
It was way back in 1953 and 1954 that Dr. Silver and Dr. Russell independently described groups of small-for-gestational-age [SGA] children whose pregnancies had been complicated by intrauterine growth restriction [IUGR]. Their common findings were short stature without catch-up growth, normal head size for age, a distinctive triangular face, low-set ears and incurving fifth fingers. These two groups of patients are now considered to have had variations of the same disorder that we now call Russell-Silver syndrome [RSS] in North America, and Silver-Russell syndrome [SRS] in Europe.
One interesting and important aspect of the Russell-Silver syndrome is its variation in phenotype. In this context, a phenotype is all the physical characteristics and abnormalities found in an individual patient that are attributed specifically to RSS. Some individuals with RSS have many traits, thus a severe phenotype, while others have very few traits, thus a mild phenotype.
When first described, RSS was NOT thought to be a genetic disorder because it recurred within families rarely, and when it did recur, its pattern of transmission failed to follow a consistent genetic mode of inheritance. More recent understandings of genetic mechanisms have led scientists to conclude that Russell-Silver syndrome is genetic, but its genetics are not simple. Scientists now know that the RSS phenotype is associated with more than one genotype.
A genotype is the status of a specific gene at a specific location on a specific chromosome. Therefore, an abnormal genotype means there has been a specific alteration, such as a deletion, duplication, insertion, substitution or imprinting error within the code of a specific gene located at a specific site in an individual's genetic code.
Since our genotype is responsible for our phenotype, abnormal genotypes result in abnormal phenotypes. If we assume several genotypes for Russell-Silver syndrome, then we should not be surprised at a variety of phenotypes. We view this as one reason for the marked variability within the group of patients considered to have RSS. But deciding which child should be considered to have RSS is not always easy. When more is known about the genetics of Russell-Silver syndrome, we will find that some patients were incorrectly included while others were incorrectly excluded.
HOW IS RUSSELL-SILVER SYNDROME DIAGNOSED?
Molecular Testing: Russell-Silver syndrome can be diagnosed with genetic testing; but negative genetic testing does not rule out a clinical diagnosis. Currently, genetic testing can be run for known causes of Russell-Silver Syndrome involving chromosomes 7 and 11. Chromosome 7 is involved in the cause of the maternal uniparental disomy of chromosome 7 [matUPD7] type of RSS, which can be either heterodisomy (rare) or isodisomy (most common).
Chromosome 11 at band p15 is involved in four different RSS mechanisms representing approximately 44-60% of RSS cases:
A) hypomethylation of the paternal imprinting control region 1 (ICR1) due to an imprinting error in up to 40-55% of cases;
B) duplication of ICR1 and ICR2 in about 2-3% of cases;
C) duplication of ICR2 in about 1% of cases;
D) mutation of CDKN1C gene in about 1% of cases.
In the rare mechanisms B through D above, which are chromosomal copy number variations (CNV), there can be a risk of transmission either to the patient’s own children or with the patient’s siblings. Genetic testing of the child’s parents will need to be completed and the results discussed with the geneticist.
There are other chromosomal abnormalities that have overlapping physical characteristics with RSS such as matUPD16, matUPD20 and matUPD14 (Temple Syndrome). As additional research is published in the future, we hope to know if these molecular abnormalities represent additional causes of Russell-Silver syndrome.
Clinical Diagnosis: These above molecular causes account for only 60-70% of RSS cases; the other 30-40% are “idiopathic” at this time, meaning the cause is unknown. For these children, doctors will generally base their diagnosis on characteristic, clinical findings that make up the RSS phenotype. It is easy to diagnose the “textbook” RSS phenotype; other cases are more difficult to classify and may require a specialist highly experienced in Russell-Silver syndrome in order to rule out another syndrome or another cause of short stature. The Netchine-Harbison RSS clinical scoring system (Azzi et al, 2015) found that the following six clinical characteristics are statistically correlated with either a positive molecular result or a clinical RSS diagnosis by an experienced specialist:
-
Small for gestational age (SGA) in birth for weight and/or length
-
Large head size for body size at BIRTH (1.5SDS difference between birth head circumference and birth weight or length)
-
Postnatal growth failure (length <-2SDS by 24mos)
-
Feeding difficulties and/or low BMI (<-2SDS at 24mos)
-
Prominent protruding forehead (age 1-3 years)
-
Body asymmetry
Until 2005, the only known molecular cause of RSS was matUPD7, which is present in less than 10% of cases. As such, many older teens and young adults may have a clinical RSS diagnosis with no confirmed molecular testing.
Please see below for more information on a free RSS screening that is offered by the MAGIC Foundation’s RSS/SGA Research & Education Fund which uses the Netchine-Harbison RSS clinical scoring system.
WHAT IS THE FREE RSS SCREENING THAT MAGIC OFFERS?
MAGIC’s RSS/SGA Research & Education Fund offers a free RSS screening using the Netchine-Harbison scoring system (Azzi et al, 2015). This screening acts as a “probability screening” – meaning based on the typical phenotype and growth patterns of RSS/SGA children, where does your child fall statistically according to the Netchine-Harbison RSS scoring system. We are also able to tell you if your child was born SGA, using the Usher & McLean standards. You can then talk with your child’s geneticist or pediatric endocrinologist who is trained in making clinical diagnoses and recommending genetic testing. If you are interested, please email rss-sgascreenings@magicfoundation.org. For more comprehensive information on diagnosing Russell-Silver syndrome, the genetics behind RSS, comprehensive explanations of the 11p15LOM and matUPD7 causes of RSS, and how these genetic causes may result in differences in RSS children, please refer to Chapter 2 and Chapter 3 of the RSS/SGA Guidebook.
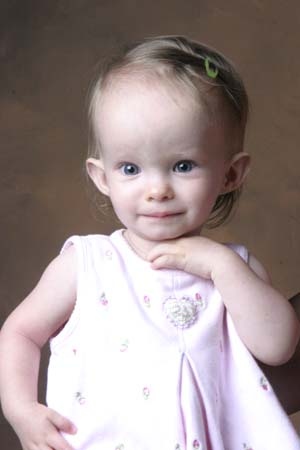
WHAT IS THE TYPICAL RUSSELL-SILVER SYNDROME PHENOTYPE (PHYSICAL CHARACTERISTICS)?
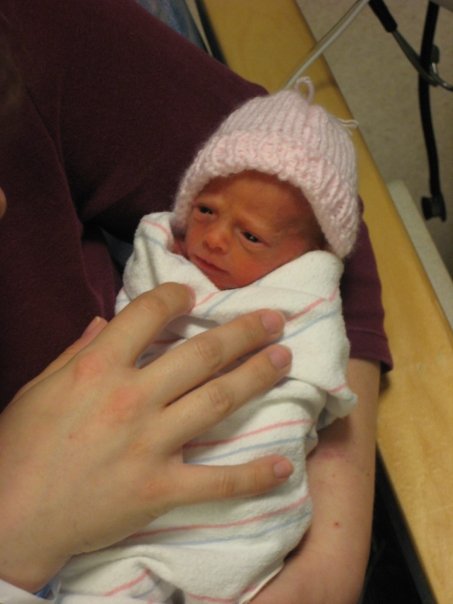
As discussed prior, the Netchine-Harbison RSS clinical scoring system found that this combination of six characteristics was able to statistically differentiate children with RSS from other IUGR/SGA children. However, this scoring system can be difficult for parents to use on their own. There is a unique combination of skeletal and craniofacial features that, to the trained eye, distinguish children with Russell-Silver syndrome from other children with growth failure. And in fact, both RSS and SGA children share many physical and functional abnormalities (functional abnormality describes a situation where an organ or a process is not working properly).
Below are two lists: the first includes the characteristics known to occur more frequently in RSS children than IUGR/SGA or the general population; the second list includes characteristics that occur more frequently in both RSS and IUGR/SGA children compared to children in the general population. It is important to note that when we refer to “SGA children” below, we are specifically referring to the small subset of children born SGA who remain small and underweight (fail to catch-up in growth). For comprehensive descriptions and information on each characteristic, including photos and incidence rates, please refer to Chapter 4 of the RSS/SGA Guidebook, pages 27-69.
Characteristics That Occur More Frequently in Russell-Silver Syndrome Children than Other Small-for-Gestational-Age Children:
(the characteristics that Azzi et al (2015) found to be statistically different between specific molecular RSS groups and SGA-non RSS subjects are marked below with an asterisk *).
-
body asymmetry – LARGE side is the “normal” side*
-
large head size for body size*
-
broad protruding forehead*
-
low-set, posteriorly rotated &/or prominent ears
-
clinodactyly (inward curving) of the 5th finger (pinky)*
-
syndactyly (webbing) of the 2nd and 3rd toes*
-
inadequate catch-up growth in first 2 years
-
persistently low weight-for-height*
-
lack of muscle mass and/or poor muscle tone*
-
hypoplastic (underdeveloped) chin & midface
-
downturned corners of mouth*
-
thin upper lip
-
high-arched palate
-
small, crowded teeth
-
unusually high-pitched voice in early years
-
café-au-lait (coffee-with-milk) birth marks
-
dimples in the posterior shoulders and hips*
-
narrow, flat feet
-
scoliosis (curved spine, associated with spinal asymmetry & accentuated by a short leg)
-
prominent heels [mUPD7 primarily]*
-
autism [mUPD7 children primarily]*
-
myoclonus dystonia (movement disorder) [mUPD7 primarily]
Characteristics of both Small-for-Gestational-Age Children and Russell-Silver Syndrome Patients:
-
lack of interest in eating
-
fasting hypoglycemia & mild metabolic acidosis
-
generalized intestinal movement abnormalities:
-
esophageal reflux resulting in movement of food up from stomach into food tube
-
delayed stomach emptying resulting in vomiting or frequent spitting up
-
slow movement of the small intestine &/or large intestine (constipation)
-
late closure of the anterior fontanel (soft spot)
-
frequent ear infections or chronic fluid in ears
-
congenital absence of the second premolars
-
delay of gross and fine motor development
-
delay of speech and oral motor development
-
kidney abnormalities
-
delayed bone age early, later fast advancement
-
early pubic hair and underarm odor (adrenarche)
-
early puberty or rarely true precocious puberty
-
classical or neurosecretory growth hormone deficiency
-
ADD and specific learning disabilities
-
blue sclera (bluish tinge in white of eye)
-
hypospadias (abnormal opening of the penis)
-
cryptorchidism (undescended testicles)
ARE THE COGNITIVE ABILITIES OF RSS CHILDREN WITHIN THE NORMAL RANGE?
An infant with RSS is generally born with normal intelligence. Learning disabilities and Attention Deficit Disorder (ADD) appear to be increased in incidence in children born IUGR/SGA for unknown reasons. Autism and similar disorders like pervasive developmental disorder (PDD) may also be increased, primarily in RSS caused by matUPD7. It is unclear whether these problems just appear to be increased in RSS/SGA, are innate to RSS/SGA, or are acquired through early malnutrition and hypoglycemia, both of which are preventable. It is very important to do everything possible to prevent hypoglycemia from occurring -- at night when it is difficult to monitor, as well as during the day especially during times of illness.
WHAT SHOULD WE DO FIRST IF WE THINK OUR CHILD HAS RUSSELL-SILVER SYNDROME?
-
Have your child's diagnosis confirmed by a doctor who is familiar with RSS/SGA patients. If you would like to request a free RSS screening from MAGIC, simply email rss@magicfoundation.org or sga@magicfoundation.org and request an RSS screening input form.
-
Make sure your child is measured carefully & frequently. KEEP YOUR OWN RECORDS. Find an endocrinologist who knows how to treat SGA children's growth failure and discuss the options.
-
Find a pediatrician who is willing to learn from experts about RSS/SGA children, and who will coordinate care and opinions with consulting specialists.
-
Get adequate calories into your child. Insufficient nutrition & low blood sugar damage the developing brain and compound the growth failure. However, due to the increased risks for insulin resistance and related health issues, it is important that an RSS child remain lean, typically with a BMI below 14.
-
Take necessary measures to prevent hypoglycemia in young RSS children. Pay special attention to the night when everyone is asleep, anytime your child is ill or not eating normally, and when your child is unusually active or stressed. Make sure to read the information on hypoglycemia on the MAGIC website in the next section.
-
Download RSS/SGA emergency medical instructions from the MAGIC website if your child is unable to eat for more than 4 hours and/or is spilling ketones in urine.
-
Treat your child his age not his size. Arrange safe, age-appropriate activities; buy age-appropriate clothes; and expect age-appropriate behavior and responsibility.
-
Watch your child's psychosocial and motor development. All states have developmental evaluation & intervention services for children younger than age 3. These programs are based on the child's needs not parental income. For children over 3 years old, the school district becomes responsible for providing these services. Take advantage of this; intervention can make a world of difference for your child!
-
Seek appropriate consultation for recurrent otitis, hypospadius, undescended testicles, leg length discrepancies, etc. But remember:
-
Only emergency surgery should be done until the child is gaining weight well.
-
A young SGA child should NEVER be fasted or kept NPO for more than 4 hours for ANY reason without glucose-running IV.
-
For surgery, IV glucose should be given during the procedure and continued in the recovery room.
WHY ARE RSS/SGA CHILDREN AT HIGH RISK FOR FASTING HYPOGLYCEMIA AND KETONURIA?
The following information is summarized and excerpted from two documents:
-
An explanatory document on RSS and hypoglycemia written by Dr. Madeleine Harbison, and
-
The book RSS/SGA - A Comprehensive Guide: Understanding Aspects of Children Diagnosed with Russell-Silver Syndrome or Born Small-for-Gestational-Age, from a chapter titled “Fasting Hypoglycemia and Ketonemia” written by Madeleine D. Harbison, M.D., Richard Stanhope, M.D. and Jennifer Salem, M.A.
This 330-page guidebook is available only through The MAGIC Foundation and includes fifteen chapters ranging from “Physical Characteristics” to “Improving Your Child’s Weight” to “Moving into Adulthood.” To receive a copy of the Guidebook, contact MAGIC at 800-3MAGIC3. Any family who becomes a new member of MAGIC will receive one FREE Guidebook. Medical professionals can also request either a free hardcopy or DVD.
Excerpts from the Guidebook....
“Since the concept of ketones is so very important for RSS parents to understand, we shall give a simple and practical overview of a very complex metabolic pathway.
Ketonemia and ketonuria normally precede hypoglycemia. Ketone body formation is associated with both a low blood sugar (hypoglycemia) and a high blood sugar, as in diabetes. In hypoglycemia, there is not enough sugar available to cells and in diabetes, the sugar cannot get into the cells. Normally, cells use sugar, (glucose), as their main fuel. When there is not enough sugar to fuel the cells, they begin to burn fat. Big fat molecules, (triglycerides) must be broken down into smaller molecule to be used as a fuel. Ketone bodies are these smaller, breakdown molecules from fat. The presence of ketones simply means that the cells are getting low on sugar and are using fat as a fuel.
Ketone bodies are weak acids. When they build-up to high levels, they cause a severe acidosis. RSS children may develop a mild acidosis from ketones but never the severe ketoacidosis seen in uncontrolled diabetes. This is because the ketones seen in RSS are associated with a LOW not a HIGH blood sugar.
Asymmetric SGA and RSS children are at an especially high risk of spilling ketones and experiencing fasting hypoglycemia because they have large brains and a small body. BRAIN CELLS cannot use fat as a fuel. The RSS child's small body can store and make only small amount of sugar. This presents a problem when they do not or cannot eat. Typically, hypoglycemia occurs during physiologic fasting, such as an overnight fast, and during episodes of illness when the child is eating less than usual, is sleeping longer than usual and is requiring more calories because of fever. Unlike the proportionate, normal child who may be able to fast for 8 to 12 hours, the fasting RSS child uses up his stored sugar within 3 to 4 hours. Upon switching to fat as a fuel, ketones appear in the blood and urine and later in the breath. Luckily they can be detected BEFORE the blood sugar drops to a dangerously low level and while the brain still has some glucose to burn. We check for ketones in the urine so that we can detect this switch over to fat burning and thus PREVENT hypoglycemia by giving sugar and complex carbohydrates.**
IMPORTANT: There are some rare, genetic metabolic disorders that prevent normal fat metabolism [SCAD, MCAD and LCAD]. They result in hypoketotic hypoglycemia. This means hypoglycemia without normal production of ketones. For these children, ketonuria does not precede hypoglycemia; thus, monitoring for ketones does not help predict their development of hypoglycemia.
As such, the safest policy is to assume that all RSS/SGA children are having nighttime hypoglycemia until proven otherwise, and steps should be taken to prevent hypoglycemia from occurring during the daytime hours. Symptoms are not hard to recognize, both ketonuria and hypoglycemia are easy to test for, and preventative measures are fairly simple to incorporate into the child care routine.
Physical Signs of Hypoglycemia:
-
waking to feed at night past early infancy
-
excessive sweating
-
extreme crankiness improved by feeding
-
difficulty waking up in the morning or extreme lethargy when ill and not able to eat
-
pallor and shakiness
-
poor coordination or odd speech
-
ketones in the urine
Testing for Ketones: Parents can easily check for ketones in their child’s urine by using urine ketone sticks or strips that are purchased over-the-counter at a pharmacy. For infants and toddlers who wear diapers, place a couple dry cotton balls in the diaper when the baby urinates. Watch for their wetting and remove the wet cotton ball immediately then squeeze it over the ketone stick or strip. Older children should urinate into a cup and the ketone stick or strip can then be dipped into the urine. The test area’s color will not change if ketones are not present. Otherwise, it will change to a varying hue of purple depending upon the concentration of ketones in the urine. Once this timed reaction is complete, the color of the test area should be compared to the color chart on the side of the bottle.
Ketone testing strips and sticks must be kept dry. All ketone measuring methods depend upon the same, moisture-sensitive chemicals in their test areas. Therefore, the strips must be kept tightly capped in a dry area, not a moist bathroom. Purchase a bottle containing the fewest tests; date the bottle when it is opened and use tests from a bottle no longer than one month after opening it.
Routine baseline testing of ketones should be done before feeding to determine if the child’s feedings are frequent enough. Typically, the longest period between feeds is overnight. It is best to measure urine ketones after the early morning diaper change. Ketone testing should be done for several consecutive days to be sure that early morning ketones are consistently negative, because of the variability in the amount of food that RSS/SGA infants and toddlers eat day to day.
If ketones are present in morning urine (or at other times), read the next section on treatment protocols for hypoglycemia and ketonemia.
Testing the Blood Sugar: Talk to your pediatrician or endocrinologist if your child is spilling ketones and/or if you feel your child needs to have his or her blood sugar tested at home. There are many brands of small, hand-held glucose monitoring devices that are ever evolving in their accuracy, ease of use and computer compatibility. There are often coupons on-line for free units, or your physician can write a prescription for a unit and supplies. Also remember, in reality, these meters are made for monitoring diabetics whose blood sugars are more frequently high than low. For this reason, it is important that RSS/SGA parents make sure that the unit they get is accurate in the lower ranges. Your physician can help facilitate this.
Preventing Hypoglycemia During Nighttime and in Routine Daily Life:
We highly encourage you to read the hypoglycemia chapter in the RSS/SGA Guidebook for comprehensive prevention and treatment information. However, here are some quick prevention tips:
-
feed frequently during the day (sometimes small snacks every 90 minutes helps)
-
keep snacks with your child at all times
-
avoid juices, sodas or foods high in refined sugar (except when these are needed for a “quick fix” for low blood sugar levels)
-
feed through a gastrosomy tube if child cannot maintain blood sugars consistently
-
utilize a “snack bag” during school hours to ensure that the child never goes more than 3-4 hours without eating
-
use cornstarch or Polycose during nighttime hours (see below)
For RSS/SGA infants and children, the nighttime hours of prolonged fasting can represent a high risk period for ketonemia which may progress to hypoglycemia, because both the child and the parents are asleep, making signs of hypoglycemia both harder to spot and frequently unobserved. It is recommended that the parent check the child’s urines for ketone evidence (see prior paragraph) anytime the child’s sleep schedule is extended. For example, if an infant goes from sleeping 4 hours at a stretch to 5 hours, test the baby’s urine with a dry cotton ball at the 5 hour mark. The ketones should be negative for a full week before the parent can assume that it is safe for the baby to fast for this longer period of 5 hours instead of 4 hours.
If an infant or child older than 8 months of age spills ketones after a nighttime fast, it is frequently possible to prevent this by adding uncooked cornstarch to his or her last feeding before bed and/or to their middle of the night feed. Giving an individual cornstarch functions like adding to the body’s stores of human starch called glycogen. The body uses the cornstarch for energy first before using the child’s limited glycogen store. An appropriate dose of cornstarch will last for close to 4 hours, and the child’s own glycogen storage will generally add another 2.5 to 4 hours – equaling a total of 6.5-8.0 hours of protection against hypoglycemia. The amount of starch needed to prevent hypoglycemia will depend upon the size of the child and his resting use of calories. Start with one level tablespoon per 4oz of milk or formula and go up as needed. The liquid needs to be stirred or shaken well as the cornstarch can settle. Three flat tablespoons is about the maximum tolerable and suspendable. The cornstarch is tasteless but can be a bit of a texture change – so we recommend slowly introducing cornstarch, beginning by mixing in only 25% of the cornstarch amount, for example, every day for a week. Then gradually increase the amount over time.
It is important to remember that below 8 months of age, infants lack the enzyme necessary to break down cornstarch. Younger infants can break down the shorter chains of glucose polymer in the product “Polycose”. Polycose, in its liquid form, may look like light corn syrup but it is not the same. Do not use corn syrup. And never give honey to a child less than 24 months of age. The bottle of Polycose should give dosing recommendations. There are rumors that Polycose may be discontinued so talk to your pediatrician.
Preventing Hypoglycemia During Times of Illness/Vomiting:
If an RSS/SGA child is out of sorts and/or seems different from usual, the parents should take the child’s temperature. If the child has a fever, the fever must be treated quickly and efficiently. A fever increases the utilization of calories, and as such, a child may use up his glycogen storage more quickly when experiencing a fever. If an RSS/SGA child is unable to eat or hold food down for 4 hours or more, he should be tested for urinary ketones. If he is spilling ketones, the parents stand alerted that their child will progress to hypoglycemia if something does not change. At this point, as early as 4 hours into the illness, the parents must do the following:
-
Bring down the fever,
-
Give a quick acting dose of carbohydrate such as glucose gel or gel cake decorating frosting,
-
Notify the child’s pediatrician,
-
Begin testing the child’s blood sugar and
-
Attempt a small feed of cool Pedialyte, flat Seven-Up or other soda.
But during bouts of severe gastrointestinal infections (such as rotovirus), the above steps simply may not be enough and the child will need gut rest and an intravenous resuscitation until the child is able to eat normally again.
Many, many parents can attest to the frustration of holding a vomiting child and having an ER doctor say that the child doesn’t need an IV because the child isn’t dehydrated. Ideally, the parent has made prior arrangements with their child’s pediatrician so that standing orders are at the local hospital. But if not, we have written a document that for medical professionals that you can use in times of illness when your child can not eat and needs an IV. We highly recommend that you print out this 2-page document and keep multiple copies in various places such as your car’s glove compartment.
Preventing Hypoglycemia in Surgical/Laboratory Procedures:
As we have said previously, RSS/SGA children generally do poorly with fasting of any amount greater than they would on a regular day. Therefore, as a rule of thumb, if a test or an anesthetic procedure requires a child to fast longer than he normally would fast at home, he will need to spend that extra time with an IV in place that delivers glucose. For surgical procedures, this will frequently mean that the child needs to be admitted and have an IV started the evening before surgery. Ideally, an IV solution with 10% solution of glucose plus electrolytes is preferred. Parents must keep in mind that frequently well-meaning surgeons will tell parents, “an IV isn’t necessary because the child is first on the morning’s surgical schedule,” and/or “the procedure is simple and will last only 30 minutes so we do not need an IV”. The reality is that emergencies can come up and a scheduled procedure can be postponed. And although a procedure may only last 30 minutes, afterwards the child may be unable or unwilling to eat so the fasting period is extended. As a result, parents must refuse to budge on this issue.
IN THE DOWNLOADS section (left side of this page) is a document to assist you in the emergency care and treatment of your RSS and/or SGA child. We encourage you to make copies of this two-page form and place one in each of your vehicles and one in your kitchen by the phone so it is readily available in the event of an emergency.”
WHAT ARE THE DIFFERENT TREATMENTS NEEDED FOR RUSSELL-SILVER SYNDROME?
An RSS child may have a variety of medical problems and special needs. And as such, you as parents will need a series of medical specialists and ancillary therapists to help you help your child be all that he or she can be. Though it is unlikely that your child will need the services of all of these specialists, listed below are the specialists/specialty services frequently involved in the care of RSS children. In the RSS/SGA Guidebook (page VI-VII), the complete responsibilities allocated to each specialty and how they apply to the RSS child are described.
It may indeed feel like you are educating everyone, including your child’s own doctors, about Russell-Silver syndrome. This to some extent will be true because most physicians caring for your child will have detailed knowledge of their specialty, but may have little or no experience with the specific and interrelated problems seen in RSS/SGA infants and children. The MAGIC Foundation can help you with this task. MAGIC will provide free to any requesting medical professional a copy of the Guidebook, and/or DVDs of select medical presentations from past MAGIC conventions. There are comprehensive chapters in the Guidebook with detailed information – for example, Chapter 5 includes 12 pages on the common gastrointestinal problems that affect RSS children and Chapter 9 includes 15 pages on ways that you can help improve the RSS child’s weight.
A Framework of Physicians and Therapies Involved in the Care of RSS/SGA Children:
● General Pediatrics ● Genetics ● Endocrinology ● Gastroenterology ● Pediatric Surgery ● Nutrition ● Speech/Feeding Therapy ● Ears, Nose & Throat (ENT) ● Dental/Orthodontics ● Urology ● Orthopedics ● Child Development ● Psychology ● Special Education Services
DO WE NEED TO IMPROVE OUR RSS CHILD’S WEIGHT? WHAT IS AN APPROPRIATE BMI FOR OUR CHILD?
In terms of actually “how” to improve caloric intake, there is simply not enough time or space here to go into this topic in depth. However, there are two extensive chapters in the RSS/SGA Guidebook on this topic – you are encouraged to read Chapter 9 “Improving Your Child’s Weight” as well as Chapter 11 “Nutrition: A Parent’s Guide.”
Improving weight can involve one or more of the following treatment methods, all of which are outlined in detail in the Guidebook: nutritional changes (both ways to increase quantity of calories as well as changing types of food that digest faster to allow for more natural hunger); identification and resolution of reflux (often asymptomatic); identification and accommodations for delayed gastric emptying; and identification and resolution of constipation (often one of the biggest causes of suppressed appetite); the use of an appetite stimulant called cyproheptadine (an antihistamine); and finally, the possibility of moving to gastrostomy tube feedings.
It is important, however, to note that it is often a balancing act with RSS/SGA children in achieving the optimal caloric intake. Abundant published research has found that IUGR/SGA children have a seemingly intrinsic insulin resistance, and that a rapid catch-up in weight gain can significantly increase their risks (both short-term and long-term) of developing health issues related to metabolic syndrome (including type 2 diabetes and other health issues). Even without rapid catch-up in weight, some research has found these same increased risks in IUGR/SGA children who have even a small amount of excess subcutaneous body fat. The goal is to have the least amount of subcutaneous body fat as possible, and for the child’s weight curve to be paralleling the normal curves not continuing to pull further down/away. Experienced RSS specialists have used the -2SDS BMI level as a “cutoff” (although this number is not always ideal due to the variance in body composition by child). A BMI that is -2SDS (or below the 3rd percentile in BMI) is about 14.0 for a girl and 14.4 for a boy. However, please know that it really is the “subcutaneous body fat” that is the problem. If the child has RSS due to 11p15 loss of methylation, typically these children have almost no muscle mass and also have body asymmetry. In these children, an ideal BMI can be as low as 12.0 or 12.5 due to the lack of muscle mass and the fact that one side of the body is smaller than the other while the BMI calculation compares the weight to the length/height of the longer-side of the body. On the other hand, a matUPD7 RSS child with no body asymmetry and high muscle mass might have an ideal BMI at 14.5 or 15.0. So the “ideal BMI” can vary.
Too often we find local doctors who lack experience with RSS, and unknowingly, have a goal to raise the child’s weight curve above the 3rd percentile. Because the RSS child’s height, without treatment, is abnormally low on the percentile curves, the weight curve will also need to remain low, even lower than the length/height curve.
On the other side of the coin, RSS/SGA children can not grow on air – meaning their length/height can be diminished if they do not consume enough calories. Especially when a child’s length percentile curve is very low as is his or her weight curve – it is useful to see if caloric repletion can improve both of these curves. If increased calories result in the child’s weight curve climbing closer to the 3rd percentile, but the length curve does not also rise, the child will simply get chubbier and this answers the question “how much of the growth failure is due to caloric insufficiency?” Typically in situations where there was caloric insufficiency, increased calories will result in the weight curve climbing up closer towards the 3rd percentile, followed by the length/height curve climbing; at some point, the length/height curve will stop climbing and begin to parallel the 3rd percentile curve (possibly still below) and the child begins to get “chubbier” (the BMI increases). This is the time when it is known that the increased calories are no longer needed for catch-up growth and your doctor may have you back off on the caloric intake.
DOES AN RSS CHILD’S DELAYED BONE AGE MEAN MORE GROWING TIME LATER?
No. Medical research has found that the delayed bone age of an RSS or SGA child does not equate to an extended amount of growing time (which is what occurs with children diagnosed with “constitutional growth delay”).
The concept of bone age is one of the most important concepts for a parent of an RSS/SGA child to understand. We strongly encourage all parents to read pages 61-66 in the RSS/SGA Guidebook for a comprehensive explanation of bone age as well as several practical examples to help a parent understand the concept and why this concept is especially important for RSS/SGA children.
Bone Age Information Highlights:
-
RSS children typically have a delayed bone age in early childhood
-
Around age 8 (sometimes earlier), this delayed bone age typically begins to advance, usually related to an early onset of adrenarche (first physical symptoms can include underarm odor and body odor).
-
By later childhood (age 9-10), the RSS child’s bone age is most often advanced (older than the child’s chronological age), which means that the child is actually losing growing time in a sense; the child will be shorter than originally predicted.
-
Bone age readings on toddlers are notoriously unreliable, with large standard errors of measurement. For infants and toddlers, an x-ray of the knee is more reliable than wrists. Once a child’s bone age is above 2 years, an X-ray of the left hand and wrist is the standard. NOTE: if your child’s left side is smaller than the right, it is recommended that X-rays are taken of BOTH wrists and readings taken of both wrists. After a child begins growth hormone therapy and/or by the age of 6, a bone age X-ray should be taken every year and compared to prior years for any advancement as compared to chronological age.
-
Once a child’s growth plates are fused, there is little to no growth potential remaining.
DO RSS CHILDREN HAVE EARLY PUBERTY?
Puberty for the general population starts on average in girls between ages 8 and 13, and in boys between ages 9 and 14. As such, the majority of RSS children do not start puberty “early” in terms of age (meaning before age 8 in girls or 9 in boys) but the starting age often is early for their height and can be at the early part of the normal age range. However, as we discussed in the prior section on bone age, it is well established that RSS children do enter adrenarche earlier than normal with a rapid bone age advancement.
It is important for parents to understand the difference between adrenarche and central puberty, the impact on both processes for children who have abnormally short height, and what treatments may be possible to slow or suppresses these processes. Complete information can be found in Chapters 4, 7 and 10 in the the RSS/SGA Guidebook. In addition, extremely early adrenarche, rapid bone age advancement, and sometimes early puberty have anecdotally been documented in MAGIC RSS children who have experienced rapid weight gain.

CAN MY RSS CHILD’S HEIGHT BE IMPROVED?
The unequivocal answer to this question is “yes”, your child’s height can be improved above the 3rd percentile and closer to the child’s midparental target height1 if you choose to use recombinant growth hormone therapy. Almost universally, research in the last two decades have found that growth hormone therapy increases childhood height for RSS/SGA children to a percentile in the “normal” range, and that GHT also increases and normalizes final adult height. And possibly more important, GHT has been found to result in many other improvements in health for RSS/SGA children. The correlated question is “how much incremental growth can we gain” and that is a much harder question to answer as there are many different variables.
In 2001, the U.S. FDA approved the use of recombinant growth hormone therapy as long-term treatment of growth failure in children who were born small-for-gestational-age and do not achieve catch-up growth by age 2. And in 2003, the European Agency for the Evaluation of Medicinal Products (EMEA) made the same approval for SGA children who had not achieved catch-up growth by age 3.
First though, the choice of whether or not to increase the RSS/SGA child’s significantly shorter stature is a personal decision that must be made by each family. Growth hormone therapy will not be the right decision for every child. Each child’s parents and endocrinologist should discuss the benefits and risks of growth hormone therapy, and discuss the various factors that impact the success of rGHT, as it relates to their specific child. The child’s endocrinologist should be able to discuss the benefits that rGHT has for RSS/SGA children in addition to increasing height, and be able to clearly articulate the risks, as well. If a child’s endocrinologist starts off any visit with the statement “rGHT isn’t proven to increase height for an RSS/SGA child”, find another endocrinogist because that statement is not true. A family may decide not to choose rGHT for their child, but they should at least have an endocrinologist who is knowledgeable about the use of rGHT in the treatment of RSS/SGA children.
The information regarding growth hormone therapy, and the impact of the pubertal process on growth, is so vast that it encompasses a full 40-page chapter in the RSS/SGA Guidebook – Chapter 10 “Improving Your Child’s Height”. There is simply no way to cover everything here, so please read the Guidebook’s lengthy and comprehensive chapter, which includes summaries of all published medical research to date. We will try and cover some important points about GHT and RSS/SGA children below.
1Midparental Target Height: Defines the adult height the child would be expected to be based on his or her parents’ adult heights. Please see the RSS/SGA Guidebook which will explain this concept and the formula.
WHAT ARE THE DIFFERENT BENEFITS AND RISKS OF GROWTH HORMONE THERAPY OTHER THAN IMPROVING HEIGHT?
As discussed before, comprehensive information about each of the benefits and risks listed below (including all of the different research studies) is in Chapter 10 of the RSS/SGA Guidebook.
Benefits of Growth Hormone Therapy for the Typical RSS/SGA Child:
-
Increased and normalized childhood height
-
Increased and normalized adult height
-
Improved appetite and weight
-
Improved BMI (body mass index)
-
Improved muscle mass
-
Improved bone mineral density
-
Improved blood pressure levels
-
Improved lipid levels
-
Improved body proportions and head circumference
-
Improved psychological and cognitive function
Possible Adverse Side Effects of GHT for RSS/SGA Children?
Large comprehensive reviews of the records of all of the major rGHT studies involving RSS/SGA children have indicated that serious adverse side effects are extremely rare. And it is critical to note that studies have concluded that GHT does not place RSS/SGA children at risk for leukemia and/or tumorigenesis (developmental of tumors). Most possible side effects have been found to be “transient”, meaning that the side effect goes away when the growth hormone treatment is stopped. These infrequent/rare side effects include the following: mild transient hypoglycemia; injection site reactions such as rashes; transient headaches, nausea and/or vomiting, usually within the first 8 weeks of starting GHT; or mild and transient edema (swelling of hands, feet or another area) early during treatment.
There are several important risks to make special note of:
-
Benign intercranial hypertension: An extremely rare risk for any child on growth hormone treatment (not just RSS/SGA), occurring in 1 in 1,000 children --- benign intercranial hypertension begins with symptoms of headaches and sometimes eyesight problems. This side effect is transient – meaning if the child develops this side effect, and the GHT is stopped, the effect goes away. This effect typically would occur during the first several months of GH treatment, not later.
-
Progression of scoliosis: Another rare side effect listed by pharmaceutical companies. There is no published data on this side effect and RSS/SGA children. It appears that GHT does not cause scoliosis to occur but rather the rapid growth acceleration common in the first year or two on GHT can cause existing scoliosis to worsen/progress.
-
Increases in IGF-I and IGFBP-3: Both levels should be monitored by the endocrinologist annually to ensure that both are kept within acceptable limits.
-
Increases in insulin levels: Insulin levels should also be monitored annually.
IS A GH STIMULATION TEST NEEDED FOR INSURANCE COVERAGE OF GHT FOR RSS/SGA CHILDREN? ARE MOST RSS/SGA CHILDREN GH DEFICIENT?
-
RSS/SGA children do NOT need to be deficient in growth hormone in order to qualify for, or benefit from, growth hormone therapy. In fact, the vast majority of RSS/SGA children demonstrate growth hormone levels in the normal range, but appear to have low normal circulating IGF-I concentrations, creating some type of GH-insufficiency rather than a classical deficiency.
-
Repeated studies have found that RSS/SGA children who are NOT growth hormone deficient grow just as much in response to GHT as those who are GH deficient.
-
Traditional GH stimulation testing (often required by insurance carriers before authorizing coverage for GHT) requires an IV and several hours of fasting, which can be dangerous for some RSS/SGA children due to the threat of hypoglycemia.
-
The child’s endocrinologist and parents should fight against a requirement of a GH stim test (unless GH-deficiency is suspected) due to the above facts: most RSS/SGA children will test normal; studies have found that the results do not predict their growth on GHT; and that the stim testing can be dangerous.
Full information including all of the cited research articles with regards to the above information is in Chapter 10 of the RSS/SGA Guidebook.
WHAT ARE SOME TIPS IN GETTING GHT COVERED BY INSURANCE?
-
In 2001, the U.S. FDA approved the use of recombinant growth hormone therapy as long-term treatment of growth failure in children who were born small-for-gestational-age (SGA) and do not achieve catch-up growth by age 2.
-
In 2003, the European Agency for the Evaluation of Medicinal Products (EMEA) made the same approval for SGA children who had not achieved catch-up growth by age 3. The FDA in the U.S. approved a maximum dose of 0.48mg/kg/week for children born SGA while EMEA approved a dose of 0.22mg/kg/week.
-
RSS children who were not born SGA may still qualify for GHT coverage under a different FDA approval ruling. In 2005, the FDA approved rGHT for a group of children under a category called “Idiopathic Short Stature.” Children with ISS are defined as those whose height is worse than -2.25 SDS from the mean (below the 3rd percentile), and whose growth velocity is so low that the child is unlikely to reach an adult height in the normal range. Talk to your endocrinologist about the ISS diagnosis possibility.
-
It is IMPORTANT that an RSS child’s medical records include the co-diagnosis of either “SGA” or “ISS” because the RSS diagnosis alone may not “qualify” a child for insurance coverage of rGHT.
-
Insurance coverage of GHT varies by carrier and policy. Coverage often requires an experienced endocrinologist in ensuring the proper paperwork and diagnosis codes are submitted. Many pharmaceutical companies have financial assistance plans as well. The MAGIC Foundation has an insurance expert on staff who can assist any member who is having insurance troubles – simply call 1-800-3MAGIC3.

WHAT ARE THE DIFFERENT FACTORS THAT CAN AFFECT OR IMPROVE THE RESPONSE TO GROWTH HORMONE THERAPY?
Various studies in the last fifteen years have identified multiple factors (also called predictors) which contribute to the success of growth hormone therapy in increasing the height of an RSS/SGA child. But keep in mind that no study has found a single factor or group of factors that accounts for even 50% of the variability, let alone 100%. Furthermore, studies have also had conflicting results. We will list below the most important factors in maximizing the success of GHT, however maximum height gain may not be the goal of every family. Please note that each factor is discussed in great detail in Chapter 10 of the RSS/SGA Guidebook.
-
Caloric intake
-
Dose level
-
Dose frequency
-
Dose compliance
-
Age at start of treatment
-
Starting height and midparental target height
-
Magnitude and duration of puberty
-
Duration of growth hormone therapy
ARE THERE OTHER HEALTH ISSUES THAT AN INDIVIDUAL WITH RSS MAY FACE IN ADULTHOOD?
Many people with Russell-Silver Syndrome (RSS) believe that once they reach their final adult height, their “RSS issues” are over. Recent research, however, shows that individuals born small-for-gestational-age (SGA) and/or diagnosed with RSS face increased risks for certain health issues in adulthood.
Listed below are the various health issues. The RSS individual may not personally experience any of these health issues, but it is important to be knowledgeable about them. You can learn more about the different health issues, the testing that should be undertaken in adulthood to monitor for these health issues, and any preventative steps you can take to reduce the risks by becoming a member of MAGIC and requesting a FREE copy of the “Transitioning to Adulthood” brochure (12-pages) as well as the free RSS/SGA Guidebook and referring to Chapter 8 “Moving Into Adulthood”.
These health risks in adulthood include the following-
Metabolic syndrome/Syndrome X, which includes:
-
Obesity
-
Insulin resistance/Type II diabetes
-
High blood pressure, especially systolic
-
Dyslipidemia
-
Coronary heart disease
-
Cerebrovascular disease
-
Polycystic ovarian syndrome (PCOS) [females]
Other possible health risks include:
-
Uterine and vaginal dysgenesis [11p15 RSS females]
-
Gonadal hypofunction/testicular cancer [males]
-
Fertility/transmission of Russell-Silver Syndrome to offspring
-
Low muscle mass/low bone mineral density
-
Autonomic dysfunction*
-
Myoclonus dystonia [mUPD7 RSS]
-
Functional issues related to the skeletal system* (body asymmetry, scoliosis/kyphosis, muscle/back pain)
* These risks are based on anecdotal evidence from the hundreds of patients seen by Dr. Madeleine Harbison, Dr. Irène Netchine and Dr. Suparana Jain.
CONCLUSION
Coping with the time-consuming special attention and services necessary to care for an RSS-SGA child can be overwhelming, especially if you try to face it alone. Good physicians often have no experience with routine needs of RSS-SGA children. Day-to-day challenges such as feeding, formulas, fitting clothes, school issues and peer pressures can be less stressful if you are in contact with other families who “have been there and done that.” Making connections between families with similar issues and facilitating sharing of information and experience is a major goal of the MAGIC Foundations RSS Division. We can put you in touch with other people who have had, and have solved, problems similar to yours.
The RSS Division has a dedicated consultant/RSS parent who is ready to communicate with you. Email ContactUs@magicfoundation.org and put RSS in the Subject line and she will respond! (Please allow for 24-48 hours for a response).
The RSS Division also has a Research & Education Fund dedicated to developing new educational tools and fostering additional research on topics related to Russell-Silver Syndrome. This group is funded 100% by family donations and each year, we choose project priorities based on parent feedback.
The MAGIC Foundation helps RSS families specifically in many ways. Following are just some of the ways MAGIC can impact your family: MAGIC continues to lobby on both the state and federal level on behalf of our families; we offer a dedicated insurance expert in the Chicago office to assist member families; we work diligently with insurance companies in an effort to maximize coverage for our families; for those children who are on growth hormone therapy, MAGIC works tirelessly with the pharmaceutical companies to ensure that financial assistance/bridge programs remain available to our qualifying families; and we provide networking between you and other RSS families as well as medical professionals that other families have recommended.
Lastly, don’t forget the educational tools we have that can help you begin your learning process about Russell-Silver Syndrome --- just go back to the Russell-Silver Syndrome home page and click on the links to the left side of the screen. The Guidebook, videos, annual convention, pamphlets, chat groups, and more are all at your fingertips. And don’t hesitate to contact the MAGIC Foundation if you have any questions at all. We are here for you.
Resources
» Download a printable version of the RSS brochure
» Download the RSS Emergency Treatment brochure
» Download a printable version of the Frequently Asked Questions when Beginning Growth Hormone Therapy brochure
» Click here for dental clinical article: Occlusal Characteristics of Individuals with GHD, ISS and RSS
» Download a printable version of the Psychosocial Impact of Short Stature on Children and Adolescents
» Click here for member benefits or to join The MAGIC Foundation. New members receive a free copy of the comprehensive RSS/SGA Resource Guidebook and/or the "Transitioning into Adulthood" brochures
» Help support continued research and education about Russell-Silver syndrome by making a tax-deductible donation to MAGIC’s RSS/SGA Research & Education Fund
» "LIKE" The MAGIC Foundation's Facebook page
» Join our closed RSS Facebook group for parents. After you request to join, please message an admin to gain access or email Dayna at rss@magicfoundation.org
"The MAGIC Foundation has been instrumental to our family in so many ways. This organization has given us access to a support network we didn't know existed. I can remember being pregnant with my son who has RSS and stumbling upon MAGIC's website when I was in the hospital on strict bedrest, talking survival rates and getting a laundry list of things that could be "wrong" with him. Nicholas was born at 37 weeks weighing only 3lbs 6oz and MAGIC was with us from the start. The info on the website gave me a starting point of what to discuss with our pediatrician as Nicholas simply wasn't growing. He was formally diagnosed with RSS and our real journey began. I can remember filling with tears as I walked into our first convention, seeing so many other kids and families who just "get it". The support and education MAGIC has given me has changed our family for the better. They have been by our side every step and being a part of the MAGIC family has really helped improve the type of care our child has received. MAGIC helps educate which leads with being better equipped to advocate for your child- this makes all the difference! Thank you MAGIC for helping our family when we need it the most!"
Lauren G., Cold Spring, KY
For assistance outside North America, visit our International Coalition to see resources in your country