Hypophosphatasia is an inherited metabolic (chemical) bone disease that results from low levels of an enzyme called alkaline phosphatase (ALP). Enzymes are proteins that act in the body's chemical reactions by breaking down other chemicals. ALP is normally present in large amounts in bone and liver. In hypophosphatasia, abnormalities in the gene that makes ALP lead to production of inactive ALP. Subsequently, several chemicals -including phosphoethanolamine, pyridoxal 5'-phosphate (a form of vitamin B6) and inorganic pyrophosphate - accumulate in the body and are found in large amounts in the blood and urine of people with Hypophosphatasia. It appears that the accumulation of inorganic pyrophosphate is the cause of the characteristic defective calcification of bones in infants and children (rickets) and in adults (osteomalacia). Nevertheless, the severity of hypophosphatasia is remarkably variable from patient-to-patient.
Symptoms & Diagnosis
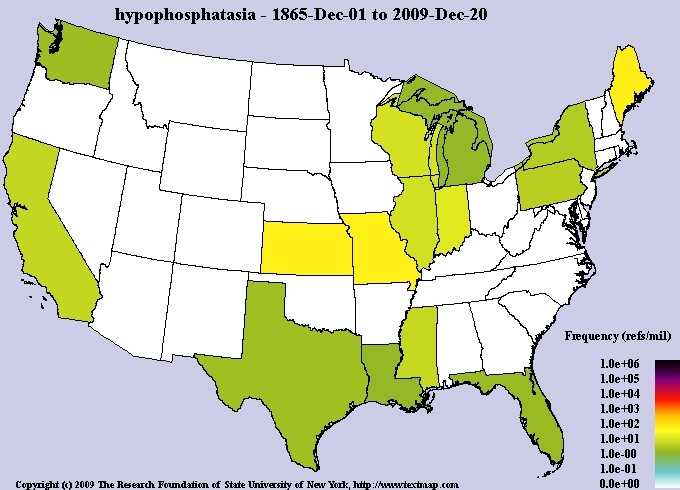 The most severely affected fail to form a skeleton in the womb and are stillborn. The most mildly affected patients may show only low levels of ALP in the blood, yet never suffer bony problems. In general, patients are categorized as having "perinatal", "childhood" or "adult" hypophosphatasia depending on the severity of the disease, which in turn is reflected by the age at which bony manifestations are first detected. Odontohypophosphatasia refers to children and adults who have only dental, but not skeletal, problems (premature loss of teeth). The x-ray changes are quite distinct to the trained eye. Similarly, the diagnosis of hypophosphatasia is largely substantiated by measuring ALP in the blood (a routine test) that is low in Hypophosphatasia. However, it is important that the doctors use appropriate age ranges for normals when interpreting an ALP level.
The most severely affected fail to form a skeleton in the womb and are stillborn. The most mildly affected patients may show only low levels of ALP in the blood, yet never suffer bony problems. In general, patients are categorized as having "perinatal", "childhood" or "adult" hypophosphatasia depending on the severity of the disease, which in turn is reflected by the age at which bony manifestations are first detected. Odontohypophosphatasia refers to children and adults who have only dental, but not skeletal, problems (premature loss of teeth). The x-ray changes are quite distinct to the trained eye. Similarly, the diagnosis of hypophosphatasia is largely substantiated by measuring ALP in the blood (a routine test) that is low in Hypophosphatasia. However, it is important that the doctors use appropriate age ranges for normals when interpreting an ALP level.
Prevalence
It has been estimated that severe forms of hypophosphatasia occur in approximately one per 100,000 live births. The more mild childhood and adult forms are probably somewhat more common. About one out of every 200 individuals in the United States may be a carrier for hypophosphatasia.
Depending on the severity of the skeletal disease, there may be deformity of the limbs and chest. Pneumonia can result if chest distortion is severe. Recurrent fractures can occur. Teeth may be lost prematurely, have wide pulp (inside) chambers, and thereby be predisposed to cavities.
Inheritance Factors
The severe perinatal and infantile forms of hypophosphatasia are inherited as autosomal recessive conditions. The patient receives one defective gene from each parent. Some more mild (childhood or adult) hypophosphatasia cases are also inherited this way. Other mild adult and odonto hypophosphatasia cases seem to be inherited in an autosomal dominant pattern (the patient gets just one defective gene, not two, transmitted from one of his/her parents). In this form, mild hypophosphatasia can occur from generation-to-generation. The perinatal form of hypophosphatasia can often be detected during pregnancy by ultrasound and by measuring ALP activity in chorionic villus samples from amniocentesis. Individuals with hypophosphatasia and parents of children with hypophosphatasia are encouraged to seek genetic counseling to explain the likelihood and severity of hypophosphatasia recurring in their families.
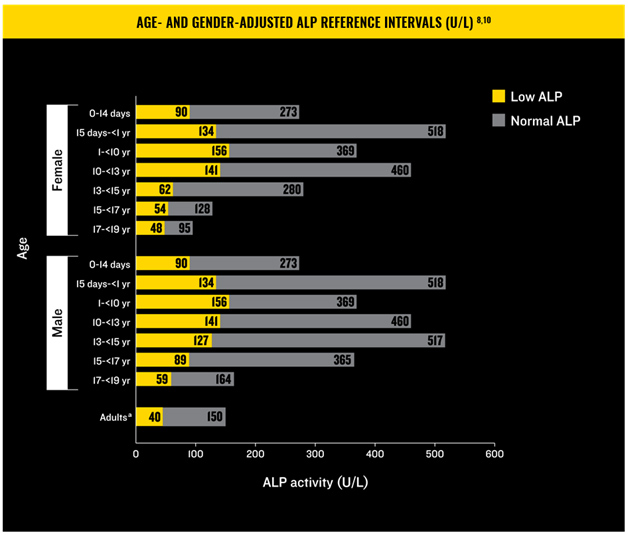
(chart can be found at hypophosphatasia.com/hcp)
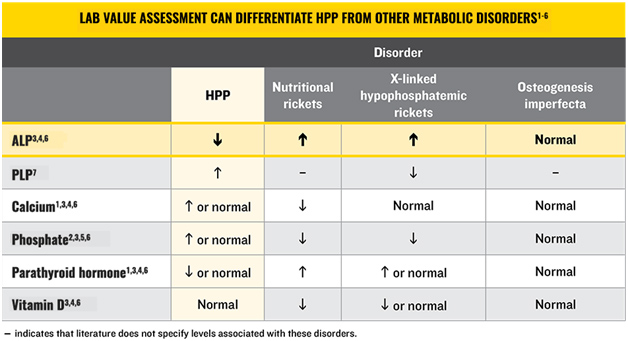
(chart can be found at hypophosphatasia.com/hcp)
Prognosis
Prognosis The outcome following a diagnosis of hypophosphatasia is very variable. In general, the earlier the diagnosis is made the more severe the skeletal manifestations. Cases detected in the womb or with severe deformity at birth almost always have a lethal outcome within days or weeks. When the diagnosis is made before six months of age, some infants have a downhill and fatal course, others survive and may even do well. When diagnosed during childhood, there can by presence or absence of skeletal deformity from underlying rickets, but premature loss of teeth (less than five years of age) is the most common manifestation. Adults may be troubled by recurrent fractures in their feet and painful, partial fractures in their thigh bones.
Treatment
For many years, treatment has been generally directed toward preventing or correcting the symptoms or complications. Procedures such as rodding, especially in adults, can be helpful with painful partial fractures in their thigh bones. Expert dental care and physical therapy are recommended. Monitoring levels such as alkaline phosphatase, B6, and calcium (which are/can be outside the limits of normal in hypophosphatasia) should be undertaken and are dependent upon how the patient is being treated.
In October 2015, a new treatment was introduced in the form of the targeted enzyme replacement therapy Strensiq (asfotase alfa). Strensiq is a tissue-nonspecific alkaline phosphatase indicated for the treatment of perinatal, infantile, and juvenile-onset hypophosphatasia (HPP), and works by replacing alkaline phosphatase. Since it’s approval, many of our families have chosen to begin this therapy. Careful consideration and consultation with a physician knowledgeable about this treatment, and the disorder hypophosphatasia itself, is recommended before starting this therapy. We have many resources available to patients and providers who are considering starting this drug to treat their hypophosphatasia. Please see the resources section below for general MAGIC information, and additional resources regarding hypophosphatasia and it’s treatment with Strensiq.
Contributing Authors:
Michael P. Whyte. M.D.
Medical Director, Metabolic Research Unit
Shriners Hospital, St. Louis, Missouri
Tracey Porreca
Hypophosphatasia, Affected Adult
Beth & Kevin's Personal Story
|
Miles was born at 9:35PM on June 8, 2017 in a suburb of Chicago, IL, and it was quickly discovered that something wasn't quite right. Miles was gasping for air and showing signs of seizures, and he was almost immediately whisked away to the NICU for what would be his home for months to come. After four, long days of tests, x-rays, and more pokes than one newborn should ever have to endure, it was discovere d that Miles's ALP levels were "immeasurable" and many of his bones were so under-mineralized, that they didn't even show up on x-ray. While we didn't have genetic confirmation at this point (that would come a few weeks later), Miles was diagnosed with Infantile Hypophosphatasia. d that Miles's ALP levels were "immeasurable" and many of his bones were so under-mineralized, that they didn't even show up on x-ray. While we didn't have genetic confirmation at this point (that would come a few weeks later), Miles was diagnosed with Infantile Hypophosphatasia.

Miles was lucky enough to be at a hospital with a geneticist who specialized in bone disorders who had heard of HPP. Without this, there’s no knowing how long it would have taken to get a correct diagnosis, if it would have ever come. Miles was also lucky enough to be born when he was, a time when there was finally a treatment for HPP. Miles received his first Strensiq injection on June 23. For the next few months, we collectively held our breaths while waiting to see if Strensiq would work.
It took about two months to start to see some development in Miles's bones. His skull that was referred to as "bone buds" when he was born was finally starting to mineralize. His ribs that looked like toothpicks on the first x-ray were showing some strength. We had no idea for these first couple of months if the enzyme replacement therapy would work. And these x-rays were the first real sign of hope because by the end of July, Miles had tired himself out from his short, shallow breathing and found himself intubated. After much discussion with his doctors, Miles had surgery to get his trach and g-tube on August 9.
 Now you'd think that with a ventilator helping him breathe and the enzyme replacement therapy helping him mineralize his bones, we would have started to see marked improvement in Miles. But that wasn't the case. Miles continued to struggle. Growing bones is apparently quite painful (I mean even the great Harry Potter thought so). After some advocating by his wonderful nurses, Miles was given a morphine drip for the pain. He was also on a number of sedative drugs. It took until November, at over 5 months old, for Miles to be weaned off of his IV drips and have his central line removed. Even after his central line was removed, it took until December for him to be weaned completely off of morphine. Now you'd think that with a ventilator helping him breathe and the enzyme replacement therapy helping him mineralize his bones, we would have started to see marked improvement in Miles. But that wasn't the case. Miles continued to struggle. Growing bones is apparently quite painful (I mean even the great Harry Potter thought so). After some advocating by his wonderful nurses, Miles was given a morphine drip for the pain. He was also on a number of sedative drugs. It took until November, at over 5 months old, for Miles to be weaned off of his IV drips and have his central line removed. Even after his central line was removed, it took until December for him to be weaned completely off of morphine.
And on January 16, 2018, after 222 days in the NICU, Miles was finally stable enough to come home. But this wasn't the end of Miles's medical adventures. Miles came home 100% ventilator and oxygen dependent. He also was fed entirely through a g-tube and never took a bottle.

For the past three and a half years, Miles has made major strides. He's learned to sit unassisted, to crawl, and then even walk on his once broken legs. He's grown strong enough to wean off the vent. And he's begun eating fully orally. But it hasn’t been all ups. Miles has lost seven teeth already, years before he “should have.” He broke his tibia when he fell down a single step while playing at a friend’s house. And last fall he had cranial vault reconstruction in the middle of a global pandemic. The biggest thing we’ve learned since Miles was born is that HPP is one heck of a rollercoaster ride. There are some crazy highs and crazy lows. But the best part of this ride is the community that’s riding with us, holding our hand as we scream in both joy and fear. And we cannot wait to see where this ride takes Miles along the way.
|