Endocrine Abnormalities
Precocious Puberty

Precocious puberty is the development of puberty before age 8 years in a girl, and before age 9 years in a boy. In McCune-Albright Syndrome, this results from peripheral precocious puberty, meaning it arises from early activation of the ovaries or testicles.
Precocious puberty is the most common endocrine abnormality in girls with McCune-Albright Syndrome. Ovarian cysts develop intermittently, resulting in breast development, vaginal bleeding, and an increased growth rate. Because ovarian cysts in McCune-Albright Syndrome usually go away on their own, surgery to remove them is NOT indicated except in very rare circumstances. Although ovarian cysts may continue into adulthood, women with MAS are often able to become pregnant and have healthy children.
Precocious puberty is less common in boys with McCune-Albright Syndrome. Symptoms in boys include an increased growth rate, pubic and axillary hair, and increased growth of the penis.
Medications are available and usually helpful for treatment of precocious puberty. A commonly used medication is letrozole, which blocks the formation of estrogen. Boys may be treated with a combination of letrozole and spironolactone, which blocks the action of testosterone.
Girls and boys with McCune-Albright Syndrome are also at risk for another type of precocious puberty, called central precocious puberty. This occurs when the area of the brain responsible for starting puberty, called the pituitary gland, turns on too early. In McCune-Albright Syndrome, this may present with signs of “breakthrough” puberty in a child who was previously well-controlled on medications. Central precocious puberty is treated with an injectable medication called leuprolide, which suppresses the pituitary gland.
Thyroid
The thyroid is a gland located in the neck, which controls many aspects of energy and growth. In McCune-Albright Syndrome, the thyroid gland may produce excess thyroid hormone, resulting in hyperthyroidism. Symptoms of hyperthyroidism can include weight loss, a fast heart rate, an increased growth rate, and hyperactivity, among others.
Hyperthyroidism in McCune-Albright Syndrome is treated with medication and surgery. Methimazole is a drug that blocks thyroid hormone production, and can be used for a few years. Most patients with McCune-Albright Syndrome and hyperthyroidism will eventually have a surgery called a thyroidectomy, where the thyroid gland is removed. After thyroidectomy patients will need thyroid hormone replacement, which is very safe and effective.
Patients with McCune-Albright Syndrome may have other thyroid abnormalities, including thyroid enlargement (called a goiter), cysts, and nodules. Patients suspected of having McCune-Albright Syndrome should have an ultrasound of the thyroid to look for these abnormalities. Patients may have abnormalities on thyroid ultrasound, but not hyperthyroidism. Some of these children may develop hyperthyroidism at a later time. For this reason, children with McCune-Albright Syndrome and abnormal thyroid ultrasounds should have thyroid hormone levels checked regularly.
Patients with McCune-Albright Syndrome who have thyroid disease may have a very slight increased risk of thyroid cancer. Because of this, radioactive iodine treatment is not recommended for treatment of hyperthyroidism in McCune-Albright Syndrome. Patients with thyroid disease should also have a doctor physically examine their thyroid gland at least once a year.
Growth Hormone
Some patients with MAS will produce high levels of growth hormone from the pituitary gland, termed growth hormone excess. The main symptom of growth hormone excess is an accelerated growth rate. Growth hormone excess may cause fibrous dysplasia to expand, and untreated growth hormone excess has been associated with a higher risk of vision loss in patients with skull disease. For this reason, it is important to screen all patients for growth hormone excess, and to carefully monitor growth in children with MAS.
Growth hormone excess may be treated with medications. Octreotide is a drug that prevents the release of growth hormone from the pituitary. Pegvisomant is a medication that blocks the action of growth hormone on its receptor. Treatments such as pituitary surgery or radiation have many side effects, and should only be used in very rare circumstances.
Cushing Syndrome
Cushing syndrome refers to excess cortisol production, a rare and potentially serious complication of McCune-Albright Syndrome. Cushing syndrome presents very early in life, during infancy or the first few years of toddlerhood. The symptoms of Cushing syndrome may vary depending on the age of the child. In infants, it may present with a low birth weight and poor weight gain. In older children, Cushing syndrome usually leads to poor growth and abnormal weight gain, especially in the face and trunk. Some patients may become severely ill, and in rare cases children with McCune-Albright Syndrome and Cushing syndrome have died.
Treatment of Cushing syndrome in McCune-Albright Syndrome is complex and depends on a number of factors, including the age of the child and the severity of illness. Patients often have surgery to remove the adrenal glands. There are also drugs which may be used to block cortisol production. In a few cases, Cushing syndrome has resolved on its own.
Phosphate Wasting
Some children with Fibrous Dysplasia / McCune-Albright Syndrome have low levels of phosphorus in the blood, called hypophosphatemia. This arises when areas of fibrous dysplasia produce excess amounts of FGF23, a hormone which causes the kidneys to lose phosphorus in the urine. Hypophosphatemia can lead to bone pain, muscles weakness, and increased fractures. Hypophosphatemia is treated with a combination of oral phosphate supplements and vitamin D.
FIBROUS DYSPLASIA
Fibrous Dysplasia (FD) is a condition where areas of the skeleton form abnormally, producing bone that is weak and deformable. Fibrous Dysplasia is extremely variable between individuals. When one bone is affected it is termed mono-ostotic Fibrous Dysplasia, and when multiple areas are affected it is termed poly-ostotic Fibrous Dysplasia.
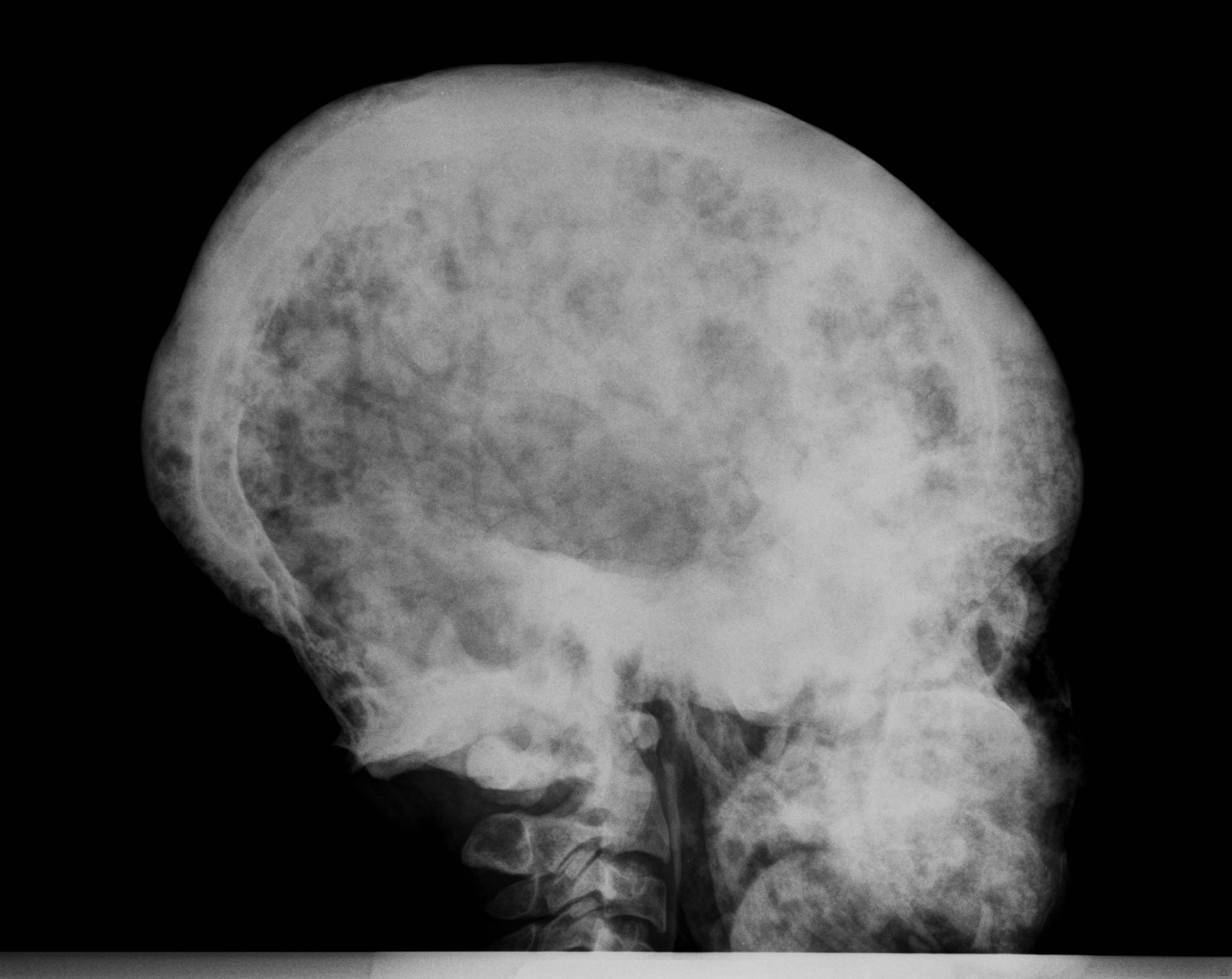
Fibrous Dysplasia can often be seen on plain X-rays. A more sensitive method of finding lesions is a bone scan, where a small amount of a radioactive tracer is injected into a vein, taken up by the Fibrous Dysplasia, and detected by a scanner. In the skull, Fibrous Dysplasia is best seen with a computed tomography (CT) scan.
Fibrous Dysplasia usually develops during early childhood. Most lesions will be visible on a bone scan by age 5 years, and most functional disability will be present by age 10 years. The bone disease usually progresses during childhood and early adulthood, and becomes less active in later adulthood.
Bone affected by Fibrous Dysplasia is weaker than normal bone, and may expand abnormally. As a result, most complications result from fracture, deformity, functional impairment, and pain. The type and severity of the complications depend on where in the skeleton Fibrous Dysplasia is located.
In the femur, Fibrous Dysplasia bone often bows to a form a shepherd’s crook deformity, which may interfere with mobility.
A potentially serious complication of Fibrous Dysplasia in the spine is scoliosis, or curvature of the spine. Most scoliosis in Fibrous Dysplasia is mild, however rarely patients have developed severe curvature, leading to compression of the lungs and even death. Fortunately surgery to correct scoliosis is very effective, and patients with Fibrous Dysplasia in the spine should be monitored regularly by an orthopedic surgeon.
In the craniofacial area (bones of the skull and face), most complications are related to Fibrous Dysplasia expansion. This may lead to facial asymmetry, and rarely loss of vision and hearing. Patients with craniofacial Fibrous Dysplasia should have a CT scan of the head to determine the extent of the disease. It is very important that all patients with craniofacial Fibrous Dysplasia have their vision and hearing tested at least once a year.
Vision loss may occur due to optic nerve damage. Fibrous Dysplasia is often located around the optic nerves, and most of the time this does not cause to vision loss. If patients develop vision changes, they usually require a surgery called an optic nerve decompression. This surgery should ONLY be performed in patients who have vision loss from Fibrous Dysplasia. It is not indicated for patients with normal vision, because there is a high risk of optic nerve damage from the surgery.
Treatment of Fibrous Dysplasia
Currently there are no medical treatments to alter the course of Fibrous Dysplasia. Surgery may be necessary to correct deformity and repair fractures. It is important to see a surgeon who is knowledgeable about the types of operations that are most effective in Fibrous Dysplasia.
Fibrous Dysplasia and Pain
Pain in Fibrous Dysplasia is common, and can occur in any location. Fibrous Dysplasia pain may be due to fracture or hypophosphatemia, or it may be related to the Fibrous Dysplasia itself. When pain develops suddenly and occurs in one specific location, the area should be evaluated for fracture. Patients should be checked for hypophosphatemia, because correction of the low phosphorus levels may alleviate the pain.
For pain that is intrinsic to Fibrous Dysplasia, there are several treatment options. Over the counter medications such as acetaminophen, ibuprofren, and naprosyn are often effective for mild to moderate pain. If pain is not controlled with these medications, a class of drugs called bisphosphonates may be effective. The bisphosphonates used most commonly to treat Fibrous Dysplasia pain are pamidronate and zoledronic acid, both given intravenously. It is important to use the lowest dose needed to control the pain. Narcotic medications should be used as a last resort for pain that is not controlled with over the counter medications or bisphosphonates.
Skin

Patients with McCune-Albright Syndrome may have areas of increased skin pigment called café-au-lait macules. The term “café-au-lait” refers to the color of the lesions in light-skinned individuals, which resembles the color of coffee with milk. In dark-skinned individuals the lesions may be more difficult to see. The pattern of the pigment distribution is unique, often starting or ending abruptly at the midline of the body. The borders tend to be rugged and irregular, and are often described as having a “coast of Maine” appearance. The macules are usually present at birth, and do not change significantly over a patient’s lifetime.
Future Directions
Research is ongoing to improve our understanding of Fibrous Dysplasia/McCune-Albright Syndrome and develop more effective treatments for patients. Some areas of interest include studying patients to better understand the Fibrous Dysplasia/McCune-Albright Syndrome disease course, developing drugs to target the abnormal Gs receptor signaling, and exploring therapies that may decrease the activity of Fibrous Dysplasia lesions. Partnership between patients, advocates, physicians, and scientists is important to help develop better treatments for patients with Fibrous Dysplasia/McCune-Albright Syndrome.
More Information
Watch this Video!
Selected References
Dumitrescu, C. E. and M. T. Collins (2008). "McCune-Albright Syndrome." Orphanet J Rare Dis. 3:12.
Collins MT, Singer FR, Eugster E (2012). "McCune-Albright Syndrome and the extraskeletal manifestations of fibrous dysplasia." Orphanet J Rare Dis. 24(7): Suppl 1:S4.
Boyce A. M., M. Glover, et al. (2012) “Optic neuropathy in McCune-Albright Syndrome: effects of early diagnosis and treatment of growth hormone excess.” J Clin Endocrinol Metab.
Brown, R. J., M. H. Kelly, et al. (2010). "Cushing syndrome in the McCune-Albright Syndrome." J Clin Endocrinol Metab 95(4): 1508-1515.
Stanton, R. P.,E. Ippolito, et al. (2012). “The surgical management of fibrous dysplasia of bone.” Orphanet J Rare Dis 7: Suppl 1:S1.
Lee J, Fitzgibbon E, Chen Y, Kim H, Lustig L, Akintoye S, Collins M, Kaban L. (2012) “Clinical guidelines for the management of craniofacial fibrous dysplasia.” Orphanet J Rare Dis 7: Suppl 1:S2
Kelly, M. H., B. Brillante, et al. (2008). "Pain in fibrous dysplasia of bone: age-related changes and the anatomical distribution of skeletal lesions." Osteoporos Int 19(1): 57-63.
Kelly M. H., B. Brillante, et al. (2005). “Physical function is impaired but quality of life preserved in patients with fibrous dysplasia of bone.” Bone 37(3):388-94.
Contributing Medical Specialists
Alison M. Boyce, MD
Pediatric Endocrinologist
Children’s National Medical Center
Washington, DC
Michael T. Collins, MD
Endocrinologist
National Institutes of Health
Bethesda, MD